HEMOGLOBINOPATÍAS
 20 Jun
20 Jun
 5 min lectura
5 min lectura
Las enfermedades hematológicas incluyen a un grupo muy amplio de condiciones como anemias, talasemias, enfermedades mieloproliferativas y metabólicas. La mayoría de estas enfermedades tienen una base genética, asociado a diversos genes, por lo que el correcto diagnóstico y manejo debe estar relacionado a un adecuado análisis genético.
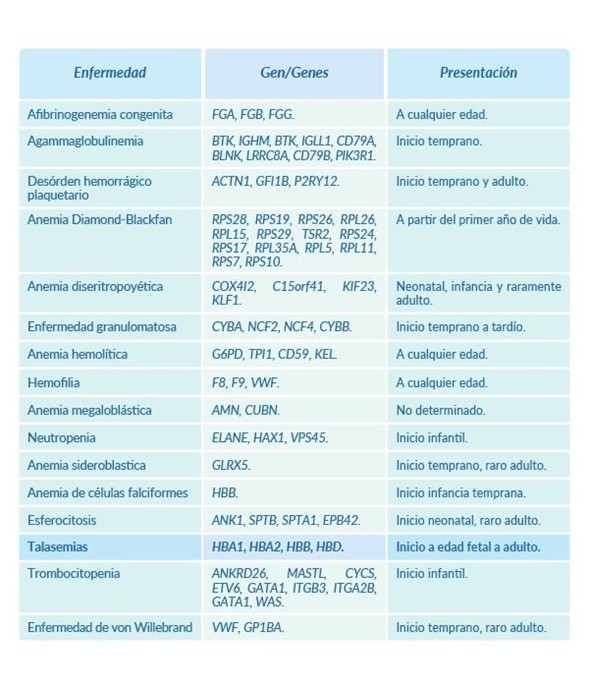
Edad de presentación y genes relacionados a enfermedades hematológicas hereditarias más prevalentes:
Las talasemias son una condición genética monogénica que corresponden a un grupo de hemoglobinopatías, con patrón de herencia autosómico recesivo, donde los pacientes poseen defectos en la síntesis de una o más cadenas polipeptídicas de la hemoglobina. Su prevalencia es alta en personas de origen Mediterráneo, sin embargo su distribución es mundial 3,4. Existen diferentes tipos de Talasemias, siendo las principales: α-talasemias y β-talasemias. La gravedad del cuadro clínico de las Talasemias varía según la configuración genética. En la α–talasemia, hay una deficiencia de síntesis de cadenas α debido a variantes patogénicas (mutaciones) en los genes HBA1 (OMIM 141800) o HBA2 (OMIM 141850), mientras que la β-talasemia se debe a variantes patogénicas en el gen HBB (OMIM 141900) lo que causa una deficiencia en la síntesis de cadenas β. 4. Hasta la fecha se han descrito más de 200 variantes patogénicas en el gen HBB.
La β-talasemia (OMIM 613985)4, implica una disminución o ausencia de la síntesis de las cadenas de β-globina, lo que resulta en anemia microcítica hipocrómica y una disminución de la HbA1. Existen tres tipos de Beta Talasemia: mayor, intermedia y menor3. Los pacientes con β-talasemia mayor (también llamada Anemia de Cooley) y β-talasemia intermedia, poseen ambas copias del gen HBB con variantes patogénicas (mutaciones bialélicas) donde puede haber una ausencia completa de la síntesis de las cadenas β o un grado variable de reducción de la síntesis de estas. Si las variantes son las mismas, se denomina al paciente como homocigoto, y si posee dos variantes diferentes se le denomina heterocigoto compuesto. Clínicamente difieren en la severidad del cuadro. Los pacientes con β-talasemia mayor presentan una anemia severa y progresiva que inicia en los primeros meses de vida, afectando su desarrollo y alimentación, asociándose otros trastornos; mientras que los pacientes con β-talasemia intermedia presentan una aneia moderada y de forma tardía, que rara vez requiere transfusión.
La β-talasemia (OMIM 613985)4, implica una disminución o ausencia de la síntesis de las cadenas de β-globina, lo que resulta en anemia microcítica hipocrómica y una disminución de la HbA1. Existen tres tipos de Beta Talasemia: mayor, intermedia y menor3. Los pacientes con β-talasemia mayor (también llamada Anemia de Cooley) y β-talasemia intermedia, poseen ambas copias del gen HBB con variantes patogénicas (mutaciones bialélicas) donde puede haber una ausencia completa de la síntesis de las cadenas β o un grado variable de reducción de la síntesis de estas. Si las variantes son las mismas, se denomina al paciente como homocigoto, y si posee dos variantes diferentes se le denomina heterocigoto compuesto. Clínicamente difieren en la severidad del cuadro. Los pacientes con β-talasemia mayor presentan una anemia severa y progresiva que inicia en los primeros meses de vida, afectando su desarrollo y alimentación, asociándose otros trastornos; mientras que los pacientes con β-talasemia intermedia presentan una aneia moderada y de forma tardía, que rara vez requiere transfusión.
El diagnóstico confirmatorio se realiza mediante el secuenciamiento del gen HBB que permitirá determinar la presencia de la(s) variante(s) en el paciente y de este modo establecer su estado de heterocigoto, homocigoto o heterocigoto compuesto3. Por ser una condición genética, el gen alterado de la β-talasemia puede heredarse a las siguientes generaciones. Los pacientes que posean ambas copias del gen HBB alterado manifestarán la β-talasemia (afectados), mientras que aquellos que posean sólo una copia del gen HBB alterado serán portadores asintomáticos. Los portadores, tienen una probabilidad del 50% de heredar la alteración a sus hijos.
Criterios de referencia al estudio genético:
- Criterios clínicos:
- Síntomas relacionados a Beta Talasemia.
- Índices de Beta Talasemia posivitos en el hemograma.
- Historia familiar positiva de Beta Talasemia.
- Variante patogénica identificada en el gen HBB en la familia.
Indices de β-talasemia en el hemograma:
Patrones de hemoglobina en β-talasemia mediante análisis cualitativo y cuantitativo de hemoglobina (cantidad y tipo de hemoglobina presente en pacientes mayores de 1 año de edad):
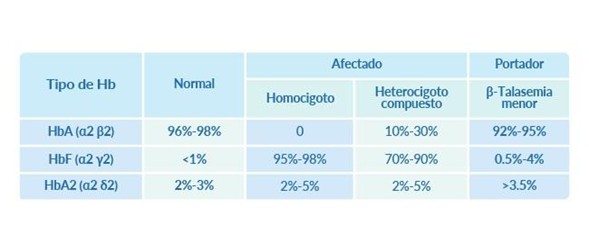
Análisis molecular del gen hbb:
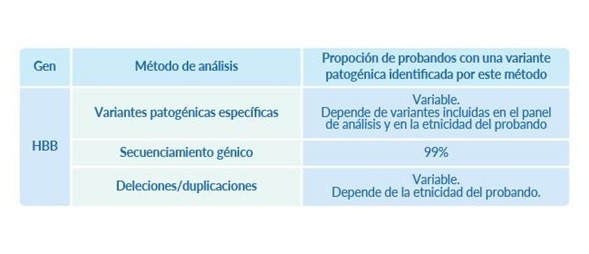
Algoritmo diagnóstico de beta talasemia:
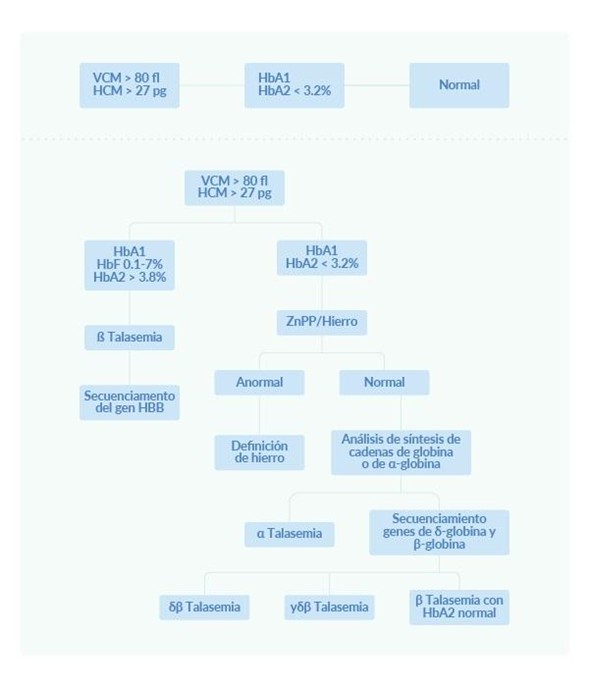
ZnPP/Hierro: Cociente entre concentraciones de protoporfirina Zinc (ZnPP) y Hierro en sangre.